Drug Review Pathways & Approval Standards
Are you sure you'd like to remove this alert? You will no longer receive email updates about this topic.
Review Pathways
Are you sure you'd like to remove this alert? You will no longer receive email updates about this topic.

Keeping Track: Two Breakthrough Oncologics, Another Antibiotic Clear US FDA Along With Pfizer’s Hemophilia B Gene Therapy
The US FDA approved Day One’s pediatric brain cancer drug Ojemda, ImmunityBio’s bladder cancer immunotherapy Anktiva, an uncomplicated UTI claim for Utility Therapeutics’ Pivya, which has a long history in Europe, and Pfizer’s hemophilia B gene therapy Beqvez.
Priority Review Vouchers: Tropical Disease Candidates Wait Years For FDA Action On Qualifying List
There are pending requests to add 13 candidates to the tropical diseases that qualify for a priority review voucher, according to a Pink Sheet analysis, as FDA Commissioner Robert Califf faces congressional pressure on the years since the list's last update.

Pink Sheet Podcast: US FDA Biosimilar Strategy, Gene Therapy Accelerated Approvals, FDA v. Partisan Politics
Pink Sheet reporter and editors discuss the impact of FDA biosimilar promotion guidance on the future of the interchangeability designation, upcoming guidance on accelerated approval for gene therapies, and partisan attacks on the agency from Capitol Hill.
Idorsia’s Novel Hypertension Pill Among Eight New Drugs To Win EMA Nod
The European Medicines Agency believes Idorsia’s Jeraygo should be approved for use in the EU at two different doses rather than just the one approved by the US Food and Drug Administration last month.

Overall Survival Is Not A Required Primary Endpoint In All Cancer Trials, US FDA Says
Seeking to clear up a ‘misconception,’ Oncology Center of Excellence officials say earlier endpoints, such as progression-free survival and overall response, continue to be useful in getting therapies to market quicker, but sponsors still should plan to systematically collect OS data to ensure there is no detriment to survival.

US FDA Gene Therapy Accelerated Approval Guidance Will Describe ‘Buckets’ Of Use Scenarios
Forthcoming guidance is expected to describe areas of “low-hanging fruit” and those that are more challenging for use of the expedited pathway, Center for Biologics Evaluation and Research Director Peter Marks said.

New EU Filings
Obecabtagene autoleucel, Autolus Therapeutics’s investigational treatment for relapsed or refractory B cell precursor acute lymphoblastic leukemia, is among the latest products that have been filed for review by the European Medicines Agency for potential EU marketing approval.

Moment Of Truth For Seven EU Filings; Cytokinetics To Make Its Case For Heart Failure Drug
The CHMP, the European Medicines Agency’s human medicines committee, will this week decide whether a range of new medicines merit being approved.
Keeping Track: Cancer Approvals From Lumisight Imaging To Adjuvant Alecensa
The US FDA’s approval of Lumicell’s optical imaging agent Lumisight makes a dozen novel approvals in 2024 for the Center for Drug Evaluation and Research.

CDER, CBER Not Seeing Hiring Slowdown Despite US FDA Warnings
FDA officials have said hiring could be slowed if an inflationary pay increase is not included in the agency budget, but CDER and CBER continue to add staff at a steady pace.

Change Is Constant For Pneumococcal Vaccines: US CDC Prepares For Merck’s V116
As Merck’s 21-valent vaccine approaches its 17 June user fee goal, the US CDC’s vaccine committee is looking at new adult age-based recommendations and bracing for a full pipeline led by GSK’s 24-valent candidate.

US FDA Expects Joint AdComm Briefing Document, Oncology Chief Says
The combined format is the new default for briefing advisory committee members, US FDA Oncology Center of Excellence Director Pazdur declared.
Drug Approval Standards
Are you sure you'd like to remove this alert? You will no longer receive email updates about this topic.
Overall Survival Is Not A Required Primary Endpoint In All Cancer Trials, US FDA Says
Seeking to clear up a ‘misconception,’ Oncology Center of Excellence officials say earlier endpoints, such as progression-free survival and overall response, continue to be useful in getting therapies to market quicker, but sponsors still should plan to systematically collect OS data to ensure there is no detriment to survival.
Preterm Birth Prevention Studies Need To Look Beyond Makena Endpoints, Experts Say
Gestational age and a binary, neonatal composite endpoint are insufficient to assess outcomes that are important to babies and families, clinicians and researchers say, suggesting development of continuous variables or scales with weighted components and a comparison of outcomes among newborns in gestational age ‘buckets.’
Minimal Residual Disease Inches Closer To Supporting Myeloma Accelerated Approvals
US FDA’s oncology advisory committee will discuss two meta-analyses supporting use of MRD as a surrogate endpoint to support accelerated approval in multiple myeloma trials.

Abecma Approval In Earlier Myeloma Carries Caution On Early Death Data
Labeling describes early death imbalance that delayed approval of Bristol Myers Squibb’s CAR-T therapy for more than three months after its user fee goal, but does not add to boxed warning; J&J’s Carvykti is due for imminent US FDA action on its own early-stage myeloma bid.

Data Integrity: US FDA Guidance Seeks To Head Off Bioavailability/Bioequivalence Study Problems
Ultimate responsibility for data integrity rests with applicant, even if study is contracted out; testing site management should build a culture of quality, says the guidance, which aims to address the high-profile data integrity problems that have plagued the generic drug industry.

Amylyx’s Relyvrio Withdrawal May Trigger More Public Pledges Based On Confirmatory Trial Data
Company makes good on vow at a September 2022 advisory committee meeting to withdraw the ALS drug if the PHOENIX trial failed. That pledge served as a backstop to FDA’s approval decision based on a single study and created a level of sponsor accountability that often is missing when postmarketing studies fail.

Don’t Stop Translational Research Once You Move To Clinic, US FDA Tells Sponsors
Drug developers often discontinue translational research as they move into late phase clinical studies – a move that can come back to haunt them, particularly if they want to rely on such data for confirmatory evidence, said Office of New Drugs Director Peter Stein.

Rare Diseases: CBER Looks To ‘Lean Into’ Accelerated Approval, Align More With CDER
US FDA biologics center officials spoke about their efforts to increase collaboration and harmonization with the drugs center, and to internally involve more review disciplines in evaluating biomarker evidence, during a Reagan-Udall Foundation meeting that weighed potential use of accelerated approval for neuronopathic mucopolysaccharidoses disorders.

Current Pathways For Rare Disease Drugs Are Not Optimal, US FDA’s Califf Says
Anticipating a ‘tsunami of therapies’ for rare diseases, commissioner says the agency will have to think of creative approaches and employ regulatory flexibility for them. FDA considers copying the oncology center’s Project Facilitate for expanded access to other diseases.

PTC’s Upstaza Takes Gene Therapy Straight To Brain (And US FDA); Translarna To Return Mid-Year
In a year full of regulatory milestones for the firm, PTC Therapeutics hopes to set some approval precedents – and basically hopes its candidates have an easier time of it than they have had before.
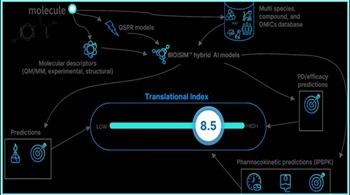
When Using Artificial Intelligence In Pharma R&D, Start With Identifying Problem To Solve
VeriSIM uses generative AI for questions such as changing a drug molecule’s chemistry and machine learning to better predict potential biological implications, says CEO Jo Varshney.

RWE: Non-Interventional Studies Must Be Able To Distinguish A True Treatment Effect, US FDA Says
Agency describes a host of issues sponsors should consider and address before pursuing observational or case-control studies to support regulatory decision-making on drug efficacy or safety; new draft guidance was issued under the FDA’s Real-World Evidence program.
You must sign in to use this functionality
Authentication.SignIn.HeadSignInHeader
Email Article
All set! This article has been sent to my@email.address.
All fields are required. For multiple recipients, separate email addresses with a semicolon.
Please Note: Only individuals with an active subscription will be able to access the full article. All other readers will be directed to the abstract and would need to subscribe.