Duke's McClellan: Changing Drug Development Policy From Outside FDA

Executive Summary
In an interview, Margolis Center for Health Policy's Mark McClellan and Gregory Daniel talk about their recent move from Brookings to Duke, the breadth and impact of their work with FDA on drug development issues, and opportunities for potential future collaborations under PDUFA VI.
Mark McClellan, the former FDA commissioner and Centers for Medicare and Medicaid Services administrator, has had a unique role in helping to inform and shape drug development and FDA oversight of medical products since leaving government service 10 years ago.
Starting in 2007, when he was named director of a new health care group within the Brookings Institution, McClellan has led a health policy team that has collaborated with FDA and other parties to better understand and develop policy solutions addressing a host of drug development and postmarketing safety challenges.
"This is kind of a different way of having impact and that's helping to identify areas where … there really are complex challenges but a lot of potential good ideas out there." – Mark McClellan
With funding from FDA, McClellan's health policy team helped get the agency's Sentinel Initiative off the ground and continues to work to develop the electronic postmarketing surveillance system as a national resource to support evidence generation. The agency also has called on McClellan and his team for assistance in supporting regulatory research pursuant to the fifth iteration of Prescription Drug User Fee Act, and to convene discussions on a host of medical policy issues impacting drug development (see graphic).
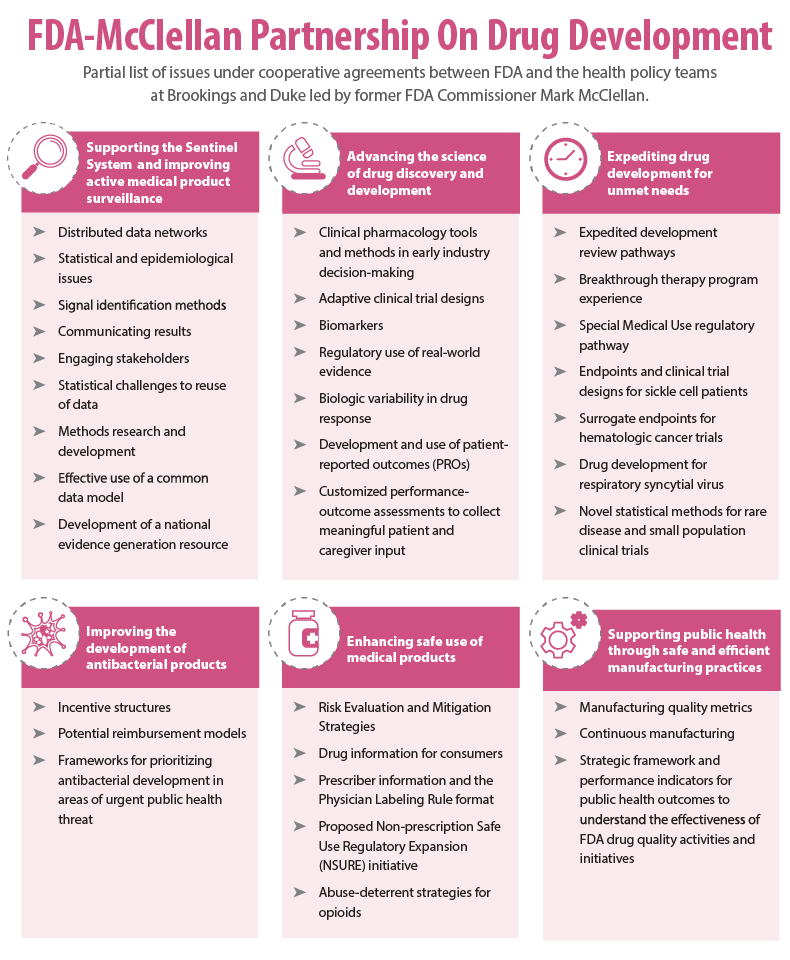
McClellan's impact on drug development and FDA policy has not been limited solely to his team's cooperative agreements with the agency. Among the notable external collaborations is the annual Conference on Clinical Cancer Research cosponsored with Friends of Cancer Research, which served as the genesis for FDA's breakthrough therapy designation program. (Also see "Expedited Approval Pathway Concept Draws Strong Support But Different Proposals" - Pink Sheet, 5 Dec, 2011.)
Mark McClellan Source: Duke-Robert J. Margolis, MD, Center for Health Policy
In January 2016, McClellan moved his health policy team from Brookings to Duke University and its newly created Margolis Center for Health Policy. There, the group is continuing to pull together public and private stakeholders to solve pressing challenges in the discovery, development, regulation and safe use of medical products to inform FDA policy. Activities include expert workshops, public meetings, white papers and pilot project development.
In a recent interview, McClellan, who is the Margolis Center's director, and Gregory Daniel, the center's deputy director who also moved over from Brookings, talked with the Pink Sheet about their move, the breadth and impact of their work with FDA to date, and opportunities for potential future collaborations with the agency under PDUFA VI. (Also see "Sponsors, FDA Reviewers To Get More Flexibility Under New User Fee Program" - Pink Sheet, 15 Jul, 2016.)
Their remarks have been edited for length, flow and clarity.
The Move To Duke
"We're still keeping a very strong presence in Washington, DC. … We moved over a lot of team, actually expanded our team in Washington since then, so we have a Duke Margolis Health Policy DC office located now at 12th and New York [Avenue] in conjunction with the Duke University DC offices, and we're going to be moving and expanding that program soon and that gives us an ability to do the same type of convening and analysis" on public policy issues in Washington.
"But Duke has offered a few other things that we didn't really have before. One is a much more extensive analytic and expert infrastructure. Duke's one of the world 's leading universities in a wide range of areas that are increasingly relevant to doing good work in improving health and health care. Aside from the Duke Clinical Research Institute, which is the world's largest clinical research enterprise, and a world class medical center at Duke, the university also has the leading business school – our main offices on campus are at the Fuqua School of Business – a leading public policy school, and a great arts and sciences program with people doing relevant work locally, regionally in the US and globally."
"Duke has a big presence in Singapore and other parts of Asia and in Europe where a lot of the issues that we're dealing with in health and health care are increasingly global. And it also has a lot of infrastructure that can be used for large collaborative projects. Whether it's very big clinical trial designs or educational programs that have reach around the world, it's all great support. And on top of that the university and thanks to the Margolis family foundation as well have made a very firm commitment to a major enhanced presence in health policy, so taking all those resources and bringing them even more to bear on public policy questions."
Difficult Issues, No Easy Answers
"What we try to do with this effort is work with FDA on an ongoing basis to identify areas where, because of complexity or lack of general knowledge, it would really help to have a neutral third-party convener of a broad range of relevant stakeholders to help make progress in understanding and thinking about next steps in these areas." – Mark McClellan
"FDA has an extensive process around developing guidances for public comment and getting public input. Our goal is to help make that public process as effective as possible, too."
"If we're going to maybe focus on a few … most important topics, maybe areas like incorporating patient-reported outcomes and more generally the patient perspective in drug development, better use of postmarketing data and better development of real-world evidence and finding ways to improve the efficiency and effectiveness of drug development."
Gregory Daniel: "All of the topics that we work on with the agency are fundamentally complex topics that take a lot of input from major stakeholder groups around the table. If they were easy issues to deal with then we probably wouldn't be working on them and the agency wouldn't really need our help."
"The patient-reported outcome instrument development and qualification also includes performance outcomes – just better ways across the board of measuring patient experiences with products … and things that really do matter to patients. Those are things that are, from a scientific perspective, really hard to measure. They're not as easy as a laboratory measure or some other physiologic measure. They do take patients' perspective and patients' understandings to really measure those kinds of outcomes. And the agency … with the patient-focused drug development program has really put a lot of emphasis into including patient perspectives throughout the drug development and review process."
"But one of the lynchpins of all of that is having valid and reliable measures to do that. There are scientific challenges to that. There are also regulatory challenges and a lot of uncertainty with drug developers and sponsors on the best ways to use those measures, the best ways to incorporate those measures and the evidence into the evidence package that they submit to the agency, and getting better clarity on when would a patient-reported outcome actually be listed in the label or the indication that could help patients or physicians make better informed decisions about the care."
"We've also seen throughout our convening that even within the agency themselves that there are lots of inconsistencies from review division to review division in how they interpret and utilize evidence on patient-reported outcomes within the regulatory process. So the agency has put a lot of effort into making that process clear and more efficient and we've been helping them through the convening process."
"One of the outputs of that has been the COA [clinical outcome assessment] compendium that the agency has published, which at least provides more clarity and visibility to sponsors and product developers about the different instruments that are out there and how best to use them." (Also see "FDA Launches Compendium Of Clinical Outcome Assessments With Eye On Patients" - Pink Sheet, 13 Jan, 2016.)
Another important issue is "the area of antibacterial drug development. That's another area where we're seeing major sort of market challenges in terms of having bacterial infections that are resistant to some of the existing antibiotics coupled with … generally a weak pipeline for drugs that really can target and address those resistant infections. The challenges for getting drugs on the market covers … scientific challenges from just understanding how bacteria evolve and how to best to respond to that evolution, to regulatory challenges in setting up the clinical trials, to major economic challenges in how those products are reimbursed and how reimbursement can be tied to better, more appropriate uses. So all of those topics have been the [focus of] some work at Brookings but then a major continuation here at Duke on identifying the market-based and policy solutions that might help incentivize drug development in that space, but also make sure that these products are being used in the most appropriate way and result in better outcomes for patients."
"Some of our activities with FDA are in fulfillment of some of their PDUFA requirements, so to hold meetings or help develop reports that stakeholders have called for as being an important priority for the agency. … A lot of this work carries into ongoing activities and forums, ongoing activities with the public around FDA's key priorities."
Gregory Daniel Source: Duke-Robert J. Margolis, MD, Center for Health Policy
"We held one of our meetings last year on kind of a review of some of the initial progress and activities under the breakthrough program, including consideration of issues like what kinds of levels of unmet need … was FDA actually using in the program as the basis for qualification and what kinds of impacts was the breakthrough designation having on the development process." (Also see "Totality Of ‘Breakthrough’ Evidence Guides FDA Designation Decisions" - Pink Sheet, 4 May, 2015.)
The meeting on breakthrough designation was a good example of "how some of the activities that we undertake with FDA can further or complement some of our other work."
Daniel: "Just on the breakthrough [designation] itself … there seemed to be a lack of clarity from companies and providers and others about what does this breakthrough therapy designation mean, what does it mean for a product that is designated a breakthrough therapy and what are the levels of evidence to get you there."
"That meeting provided a lot of clarity … because it actually got the FDA to think out loud … in terms of … particular example cases that we walked through during the meeting and what in their mind led to the level that was needed for breakthrough versus what was missing in some of the products that we used as case examples that didn't get the breakthrough designation."
"The antibiotics work and other work that we've done related to real-world evidence development does lead into areas that we work on with the agency but are areas that we continue to support within our center with other collaborators, and it's a great way to make sure the topics don't just stop with one event or one paper but that we can carry forward."
McClellan: "Obviously the center's covering a broad range of topics. Getting back to the specifics for FDA, just for example in this area of improving and making more efficient the development and the ability of effective drugs for unmet needs, FDA currently has mandates under FDASIA Section 901 to explore better uses of accelerated approval and fast track, and as a part of that some of our recent and upcoming events are helping to bring some technical expertise and different perspectives to bear on how to do it, something that will then inform potentially FDA's further activities, guidance that it might put out for public comment and inform and improve the ability of a wide range of stakeholders to help work with FDA to address these challenges."
"For example, we're working on surrogate endpoints for hematologic cancer trials, such as minimal residual disease." (Also see "Cancer Trial Endpoints: Minimal Residual Disease Eyed As Surrogate" - Pink Sheet, 20 Sep, 2016.)
"We recently had a meeting on advancing drug development for respiratory syncytial virus, RSV, which is the leading cause of lower respiratory infections in infants and children and has not seen a whole lot of progress and development recently. There are issues around how you design trials in this population. Also some work coming up on novel methods for very small population trials, more targeted therapies. "
Real-World Evidence, Biomarker Work To Continue Under PDUFA VI
Commissioner Robert Califf and Center for Drug Evaluation and Research Director Janet Woodcock "have both publicly stated the importance of making sure that we're utilizing to the best abilities that we can and making more efficient the evidence development process itself, recognizing that traditional clinical trials are great in what they're intended to do, but they're also very costly and burdensome to the health care system."
"The idea is, can routinely collected health information like through electronic medical records, claims data, patient registries, etc., can that also supplement and inform clinical trials and supplement the evidence that's brought to bear? So we started working with the agency on that topic, we formed a planning group, we had an expert workshop and a public event." (Also see "Real-World Evidence: Efficacy Assessments Await FDA Clarity, Pilot Projects" - Pink Sheet, 14 Mar, 2016.)
"We continue to work on refining the ideas that have come out in those convenings, working on better ways to collect evidence and use evidence and on a series of papers that … can serve as great inputs to what the agency might be held accountable for through PDUFA VI, which might be guidances and other things for how to best use real-world evidence development. So that's a project that seems to have a lot of support and more importantly input from payers and patients and manufacturers and other electronic health record experts, etc."
"Another topic that we continue to work on is that push for better incorporating the patient voice into the benefit/risk framework and that starts with understanding needs that patients have, the experiences that they have with the diseases, and getting a better sense of what really matters to them in terms of effectiveness. … Having the tools and infrastructure to actually be able to collect that information and include it in the trials and in the evidence development activities that are happening is another thing, and getting experts in the science of collecting patient information and working through approaches to best incorporate that into the regulatory process is something that we continue to do. We did that more with patient-reported outcomes. We're currently working on … performance outcomes that are important for patients and important in the drug development process. Performance outcomes are things that measure cognitive, sensory and physical function, and those are also hard to collect and need some concerted effort on the science and the ability to collect that information."
"Other areas that are critically important are areas that help to improve the efficiency of drug development itself. The time and costs for getting a drug to market are substantial … and getting a better sense on how drug development tools like biomarkers could be more used and more useful in getting important evidence developed rather than waiting really long for the true clinical outcome to develop."
"The science is one thing, but the evidentiary criteria for using biomarkers like surrogate endpoints or even predictive and prognostic biomarkers that help adaptive clinical trials early to help identify patients most likely to respond to therapy and including that in the clinical trial process itself is a challenging topic and there's not a lot of clarity on how best to do that. It takes scientific experts, trial design experts and others to really think through the issues and help provide inputs to the agency in developing guidance and things like that."
"So the patient voice in drug development, real-world evidence use, biomarkers and drug development tool qualification – those are all really important topics that we've been working on and I think will continue to be important topics in helping to improve the efficiency of drug development and keep it patient-centered."
"The patient voice in drug development, real-world evidence use, biomarkers and drug development tool qualification – are all really important topics that we've been working on and I think will continue to be important topics in helping to improve the efficiency of drug development and keep it patient-centered." – Gregory Daniel
"But one of the things that I noticed then and that sort of informs our work now is that FDA activities haven't historically gotten as much attention as they might in the scientific community and the consumer and patient community."
"This is kind of a different way of having impact and that's helping to identify areas where … there really are complex challenges but a lot of potential good ideas out there and helping make sure that those get into broader discussion now with the agency, with the public, with stakeholders."
"A big goal of PDUFA every time around [has been] about improving the regulatory process and improving drug development, but that turns out to be hard, so I see this as kind of a different role in supporting the development of better ideas, supporting a better system that can help FDA achieve its mission."