Biosimilars And The Need For Real-World Evidence
This article was originally published in SRA
The success of biosimilars will depend on a range of factors, including local policies on interchangeability, harmonization of naming conventions, reimbursement decisions, and physician adoption. Real-world evidence will help to support data generated by clinical trials and fill the evidence gap for doctors, patients and payers, say Jaclyn Bosco and Nancy Dreyer.
With increasing biologic patent expiries, availability of biosimilars is expected to increase. While the lower cost of these biologics may entice some payers, biosimilar manufacturers face a sophisticated marketplace. Relevant real-world clinical evidence will be needed to support claims of safety, effectiveness and value to satisfy not only regulatory concerns but also those of clinicians, patients and health systems.
This article discusses the role of real-world evidence generation for demonstrating safety and effectiveness, evaluating treatment heterogeneity and identifying any delayed risks and benefits related to biosimilars. Specifically, the article examines considerations involved in framing research questions, designing studies, acquiring data, tracking treatment exposure and accounting for confounding factors.
Biologics are products that are developed through biologic synthesis in living organisms. They are used in vaccines, blood or blood components, gene and cell therapies, tissues and therapeutic proteins, including monoclonal antibodies. Advances in biologics have provided successful treatments for cancer, rheumatologic diseases and other conditions but these products are far more costly than small molecule drugs. For example, the costs of protein-based therapeutic products used to treat diabetes and anemia as well as more rare conditions range from $15,000 to $150,000 per year1.
The promise of biosimilars is to provide cost savings, increase patient access, and promote innovation2. Biologic medicines accounted for 27% (€36bn) of drug spending in Europe in 20113 and for 28% ($92bn) in the US in 20134, although they accounted for less than 1% of all prescriptions dispensed in the US.
While biosimilars are intended to be more affordable to patients than the branded biologic, the cost-savings are not as great as for small-molecule generics because of the complexity of synthesizing biosimilars in living organisms. Of the 21 biosimilars approved by the European Medicines Agency since 2006, 19 are currently marketed5 and studies have shown average discounts of at least 25% compared with the originator product6. Recently, discounts up to 70% for biosimilar infliximab have been seen in Norway7. However, deep discounts may not be widely offered for complex biologics like infliximab due to the greater complexity in production. With many of the top-selling biologic patent expiries on the horizon, the availability and affordability of biosimilars is expected to dramatically increase across the globe.
A biosimilar is a highly similar version of an already authorized biologic (originator or reference product). Generally, in addition to comparative Phase I PK/PD data, a pivotal Phase III trial with head-to-head comparisons in the most sensitive indication to demonstrate bioequivalence is required by regulators. After demonstrating bioequivalence, it is likely that regulators will grant extrapolations to all other approved indications as the reference product based on scientific justifications for the mechanism of action8.9. A risk management or pharmacovigilance plan is almost always required and may mimic the reference product’s pharmacovigilance measures, and can include additional data collection for the biosimilar. Post-marketing studies may also be required given the uncertainty about biosimilar purity and real-world safety and effectiveness.
Real-World Evidence Generation
Real-world evidence generation can span the clinical development pathway (see Figure 1) and fill the knowledge gap for many stakeholders. The ideal approach is to generate real-world evidence with multiple stakeholders in mind at the outset, so that the research can serve more than one purpose.
Figure 1: Early engagement to integrate development pathways
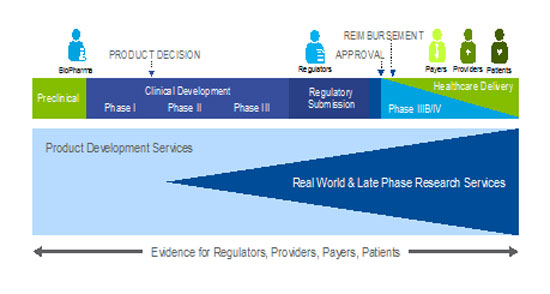
Post-approval stakeholder research priorities can be broadly categorized into three major areas:
- Safety and effectiveness.
- Heterogeneity of treatment effect.
- Delayed risks and benefits.
Once regulatory approval is obtained, regulatory agencies tend to focus mainly on safety issues. In contrast, clinicians and patients are interested in both effectiveness and safety, as well as how the risks and benefits of one treatment compare with another. In practice, these research priorities can be evaluated through biosimilar-only studies, or using broader data collection strategies to facilitate comparisons with the originator or other marketed products for various treatments and sensitive subgroups.
Payers’ interests are similar to those of healthcare providers and patients, but economic risk-benefit considerations are paramount, as is information that gives them insights into medical care, especially for high cost subgroups. The time frames for these economic considerations depend, in part, on the local health care system. For example, in contrast to the National Health Service in the UK which provides coverage from "cradle to grave", in the US the average duration of coverage by private health insurance for an adult member under age 65 is about 2.5 years; thus delayed risks and benefits may be less important to stakeholders in markets that do not provide lifetime coverage. Economic considerations are also important to patients and healthcare providers, since in some countries, patients directly bear the costs of pharmacotherapy (e.g., China and India). These economic considerations must be balanced with benefits achieved in broader population access to these life-altering treatments.
Methodological Considerations
Careful design and implementation of real-world studies will be key to generating high-quality evidence to support decision-making around biosimilars. Methodological considerations relate to the evolving regulatory and policy landscape on issues such as non-harmonized naming conventions, interchangeability and automatic substitution, as well as decisions regarding reimbursement, clinical guideline recommendations, and physician knowledge and adoption of biosimilars – all of which can vary from country to country and within different states within a country.
Different naming conventions for biosimilars have been adopted by various jurisdictions. For example, the EMA has licensed all of its biosimilars under the same International Nonproprietary Name (INN) as the originator product. However, the US Food and Drug Administration approved the first US biosimilar with a temporary INN combined with a company-specific suffix of "filgrastim-sndz," to distinguish it from the originator10,11. On Aug. 27, the FDA issued a draft guidance document and a proposed rule under which the agency wants all biologics, including biosimilars, to carry a unique four-letter suffix as part of the nonproprietary names (INNs) of the active drug substance. The suffix is supposed to be devoid of meaning and not promotional, among other things12-14. Other countries such as South Korea use only the proprietary name15. In an effort to harmonize naming of biosimilars, the World Health Organization also has proposed including a four-letter suffix assigned at random as a biologic qualifier (BQ) for the naming of all biologics16,17. In this proposal, a biosimilar product may have more than one BQ for the same biologic product if varying regulatory authorities have different comparability assessments.
Legislation around interchangeability and automatic substitution also varies by regulatory authority. In the EU, for example, interchangeability is not part of the EMA’s Committee for Medicinal Products for Human Use (CHMP) remit but is the responsibility of the national competent authorities18. In the US the FDA can approve a biosimilar as "interchangeable" or "not interchangeable"19, but individual state laws govern the ability of a pharmacist to make substitutions for a branded product. Currently, the FDA is continuing to consider what type of scientific evidence is needed to support an interchangeability approval.
Some argue that the safety and effectiveness of a biosimilar are known at the time of its approval because it receives marketing authorization only after demonstrating that it is highly similar to the originator product. However, others claim that there may be differences in a biosimilar that could generate increased immunogenicity or other safety events due to minor modifications to manufacturing processes, or simply result in product drift20. Different naming conventions and policies around interchangeability and automatic substitution will affect pharmacovigilance reporting and the ability to track exactly which medication a patient has received in routine clinical practice.
The appropriate study design for real-world evidence generation will depend on the research needs as well as the uptake of the biosimilar in the market of interest. Study designs and approaches can include, but are not limited to, prospective non-interventional studies with de novo data collection, database studies using existing data sources, and minimally interventional studies. The availability of existing databases in countries of interest will vary, and it is unlikely that any database will contain all the information needed on interchangeability and substitution, clinical characteristics, and confirmation that the prescribed drug was the administered drug. De novo data collection would enable prospective follow-up, capturing the brand at the time of dosing for products that are administered, allowing also for the inclusion of patient-reported and/or physician-reported data collection. Supplementing prospective data collection with pharmacy and other data may be a suitable option, depending on locality and data availability.
Information on biosimilar treatments should take into account the chronic use of a product, with start and stop dates, dose escalations or reductions, and treatment interruptions or "holidays." Drug switching also needs to be accounted for, since product switches may occur several times during follow-up. Moreover, the induction time for safety events of biologics may not be fully understood, making it hard to determine whether a current therapy (biosimilar) is to blame, or the originator that was taken for years prior to the biosimilar, thus emphasizing the need to distinguish between each product that has been administered.
For a comparative study, naming conventions and interchangeability/substitution affect the feasibility of identificying and selecting appropriate comparators and are dependent on the target market(s). Hypothetically, in a market where the biosimilar is indistinguishable from the branded product because it was approved with an identical INN and pharmacy-level substitution is allowed, the biosimilar and comparator group would only be identified by the proprietary name. However, the proprietary name may not be routinely recorded in the medical record or distinguishable in available data. The choice of the comparator will depend on product availability and clinical practice in the local market and can be the originator product, other biosimilars of the same originator biologic, or other treatment modalities.
Given that biosimilars would be approved with the same labeled indications and dosing administration as the originator, little to no mixing of effects (confounding) due to patient-level factors and prognostic characteristics would be expected since prescribing would depend largely on market factors and not as much on patient factors. Physician and patient preference, product supply and insurance coverage are examples of potential confounders, because it becomes difficult to extricate true treatment effects from these extraneous factors. While confounding is a concern in all non-interventional studies, in real-world studies of biosimilars patients are being treated for chronic and severe conditions and often they receive other treatments in addition to the biosimilar or biologic of interest, e.g., cancer patients receiving pegfilgrastim are also receiving chemotherapy, which makes it difficult to attribute a safety event to the correct product.
Channeling bias happens when drugs with similar therapeutic indications are prescribed to groups of patients with prognostic differences. For example, physicians may prescribe the newly approved biosimilar more often to patients who are biologically naïve and not to patients who have been stable on the original biologic. With the uncertainty about whether biosimilars are as effective and safe, there could be a strong influence by physician prescribing preference that may cloud the true effect between the biosimilar and long-term outcomes.
Conclusion
The success of biosimilars depends on many considerations that differ from those relating to small-molecule products and their generic versions. These considerations will depend on the markets of interest given the variability in local legislation and policy on issues such as interchangeability and automatic substitution, non-harmonized naming conventions as well as decisions regarding reimbursement, clinical guideline recommendations, and physician knowledge and adoption of biosimilars. Real-world studies can be used to generate clinical evidence that can supplement pivotal Phase III data and build physician confidence in biosimilar safety and effectiveness, payer assurance of value, and patient understanding of appropriate treatment options. However, accurate identification of the biosimilar versus the originator product, and the conditions for its use, will be critical to successfully attributing safety and effectiveness outcomes to the correct product.
References
1. Dudzinski DM, Kesselheim AS, Scientific and legal viability of follow-on protein drugs, N Engl J Med, 2008;358:8:843-848
2. Sarpatwari A, Avorn J, Kesselheim AS, Progress and hurdles for follow-on biologics, New Engl J Med, 2015, 372;25:2380-2382
3. IMS Institute for Healthcare Informatics, Assessing biosimilar uptake and competition in European markets, October 2014, www.imshealth.com/imshealth/Global/Content/Corporate/IMS%20Health%20Institute/Insights/Assessing_biosimilar_uptake_and_competition_in_European_markets.pdf
4. IMS Institute for Healthcare Informatics, October 2014, Medicine Use and Shifting Costs of Healthcare: A Review of the use of Medicines in the United States in 2013
5. European Medicines Agency, European public assessment reports, website accessed July 27, 2015, www.ema.europa.eu/ema/index.jsp?curl=pages%2Fmedicines%2Flanding%2Fepar_search.jsp&mid=WC0b01ac058001d124&searchTab=searchByAuthType&alreadyLoaded=true&isNewQuery=true&status=Authorised&status=Withdrawn&status=Suspended&status=Refused&keyword=Enter+keywords&searchType=name&taxonomyPath=&treeNumber=&searchGenericType=biosimilars&genericsKeywordSearch=Submit
6. Grabowski, H et al, Biosimilar Competition: Lessons from Europe, Nature Reviews Drug Discovery, 13(2), 2014, 99-100
7. GaBI Online, Huge discount on biosimilar infliximab in Norway, March 13, 2015, www.gabionline.net/Biosimilars/General/Huge-discount-on-biosimilar-infliximab-in-Norway
8. EMA, Guideline for Similar Biological Medicinal Products, CHMP/437/04 Rev 1, Oct.21, 2014, www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf
9. US FDA guidance, Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product, May 2014, www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM397017.pdfn%20351(i)(2)%20of%20the%20PHS%20Act
10. FDA press release, March 6, 2015, www.fda.gov/newsevents/newsroom/pressannouncements/ucm436648.htm
11. Novartis/Sandoz make US history with 1st biosimilar approval, Scrip Regulatory Affairs, March 9, 2015
12. 4-Letter Word: US FDA's Biosimilars/Biologics Distinction, Scrip Regulatory Affairs, Aug. 28 2015
13. FDA draft guidance, Nonproprietary Naming of Biological Products, August 2015, www.pharmamedtechbi.com/~/media/Supporting%20Documents/The%20Pink%20Sheet%20DAILY/2015/August/Biosimilar%20naming%20draft%20guidance%2082715.pdf
14. Federal Register, Proposed rule, Designation of Official Names and Proper Names for Certain Biological Products, Aug. 28, 2015, www.pharmamedtechbi.com/~/media/Supporting%20Documents/The%20Pink%20Sheet%20DAILY/2015/August/Biosimilar%20naming%20proposed%20rule%2082715.pdf
15. Debate over naming of biosimilars intensifies ahead of WHO meeting, The Pharmaceutical Journal, June 13, 2015, 294(7866),
16. EU stands firm on biosimilar naming as WHO pursues qualifier plan, Scrip Regulatory Affairs, April 27, 2015
17. WHO, Biological Qualifier: An INN Proposal (draft), Programme on International Nonproprietary Names, INN Working Doc. 14.342, Revised draft June 2015, www.who.int/medicines/services/inn/bq_innproposal201506.pdf.pdf?ua=1
18. EMA Procedural advice for users of the Centralised Procedure for Similar Biological Medicinal Products applications, EMA/940451/2011, October 2014, www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2012/04/WC500125166.pdf
19. See Reference 9
20. Ramanan S, Grampp G, Drift, evolution, and divergence in biologics and biosimilars manufacturing, BioDrugs, 2014 Aug;28(4):363-72. doi: 10.1007/s40259-014-0088-z
Jaclyn L. F. Bosco, PhD, MPH, is Director of Epidemiology and Outcomes Research, Real-World & Late Phase Research, at global biopharmaceutical service provider Quintiles. Nancy A. Dreyer, PhD, MPH, is Global Chief of Scientific Affairs, Sr. VP Real-World & Late Phase Research, at the same company. Emails: [email protected] and [email protected].