Preparing for change - again - with the XEVMPD
This article was originally published in SRA
The EMA’s update of requirements on submitting data to the extended Eudravigilance medicinal product dictionary brings significant changes. Andrew Marr explains how the newly issued requirements will affect pharmaceutical companies in 2014 and beyond.
The European Medicines Agency has amended the requirements on the data pharmaceutical companies must submit to the extended Eudravigilance Medicinal Product Dictionary, or XEVMPD1. The amended requirements, published in a guidance document on 31 January, will have a significant impact on all product records submitted so far and on how they are to be maintained in future.
The new requirements are being introduced to address a range of issues that include:
- new information required to support the assignment of pharmacovigilance fees;
- improvements to the quality of the data being submitted;
- modification of controlled vocabularies;
- provision of local language summary of product characteristics (SmPCs) for mutual recognition procedure (MRP) products and decentralized procedure (DCP) products; and
- timelines and processes for maintenance of the data.
The requirements will start to apply from 16 June this year, and by the end of December all companies must have updated their records in accordance with the guidance and to reflect any change to their data that has been driven by regulatory activities since they were submitted initially. Once the records have been updated, companies must maintain the data within a 30 calendar day timeframe of any subsequent change. After it has conducted quality control checks on re-submitted data, the EMA from January 2015 will begin to utilize the data in a variety of business processes including in pharmacovigilance assessments. These processes will continue until the implementation of the ISO standards for Identification of Medicinal Products (IDMP), which is scheduled by legislation to be implemented in 2016 (see Table 1)2.
Table 1. Key milestones in the timelines for updating XEVMPD records and the implementation of ISO IDMP standards
| Date | Milestone |
| Now – 16 June 2014 | Preparation |
| 16 June 2014 | Start of re-submission window |
| 31 December 2014 | End of re-submission window |
| From 16 June 2014 and not later than 1 January 2015 | Maintain XEVMPD records once updated |
| 1 January 2015 | EMA begins to use the data |
| 2016 | Implementation of ISO IDMP* |
* Article 40 of the Pharmacovigilance implementing measures states 1 July 2016 as the date
Several documents have been posted by the EMA and more are to follow. Revised training and support will be forthcoming in the run-up to June 2014. Documents issued so far are numerous, the key ones being:
- a press release – which defines the reasons for change and the timescales involved and which emphasizes that it is a legally binding requirement for market authorization holders (MAHs) to submit the data and keep it up to date;
- a legal notice – which expands upon the responsibilities, requirements and timelines 3;
- reporting requirements for MAHs – which provides more details on the phases of activities and expectations of MAHs4;
- revised guidance (Chapter 3.I – technical guidance (including a revised XEVPRM, or extended Eudravigilance Product Report Message, schema), Chapter 3.II – user guidance 5), and
- summary of changes to the XEVPRM schema and EV Web6.
In March, it is expected that a document will be released that describes the quality control methodology that should be applied by companies and the EMA.
Assigning fees for pharmacovigilance
Under the draft pharmacovigilance fees legislation7, the EMA will assume responsibilities to assign and collect fees for pharmacovigilance. The legislation has not yet been agreed by the European Parliament, which is set to consider it in April, and so it is potentially subject to change. However, the proposals mean that the XEVMPD database is to be used as a basis for managing the fees and that the fees are subject to differences dependent upon the size of the company and the types of products. The EMA needs to be ready to support this from January 2015 and thus is implementing changes in time to meet the deadline. Of course, should the Parliament modify the proposals, there may need to be further changes.
For each organization (MAH), additional information is required on whether or not they are classified as a small or medium-sized enterprise (SME) and, if so, their SME number. For each authorized medicinal product (AMP), additional information is to be provided on the authorized pharmaceutical dosage form (as distinct from the administrable pharmaceutical dosage form that has already been provided), the legal basis of approval for the product and the medicinal product type. These latter two are also to help the EMA determine which products fall within Article 57(2) of the legislation as there are records that have been submitted for products that are not covered and this affects the fees to be assigned. Products that pre-date medicinal product legislative changes in 2001 will need to have assigned the legal basis that would be used if the product were to be registered today.
The impact of this is that every organization and AMP record must, at a minimum, be updated with this information. Within the EV Web tool, the EMA hopes to provide a means by which records can be updated as a batch but it has not yet announced this. Companies that manage their own data and feed these through the EV Gateway will need to update their records internally and then submit the changes.
It should be noted that once the new schema is implemented in June it will no longer be possible to submit records using the old schema. This does not affect EV Web users but will affect EV Gateway users who will require new versions of their software to handle the new data fields and schema before they can submit any new or updated records.
Poor quality data
To understand the drivers for some of the changes to the guidance, it is necessary to look at what has occurred since the submission of the initial data that was targeted for completion in July 2012. Statistics made available by the EMA show that by September 2012, approximately 240,000 product records had been submitted and this had risen to approximately 450,000 records by October 2013, at which point the agency was still receiving 5,000-10,000 new product records per month.
The data submitted has not been quality checked or used in support of any processes at the EMA. The agency did conduct two small-scale pilot assessments of the quality of the data but the results were only reported in May 2013, well after the submission of most records. These pilot assessments have shown an unacceptably high error rate. In the second pilot of 85 products/365 presentations, the error rate against the EMA’s validation criteria was 18.5%. The number of products with errors was 84% (see Figure 1).
Figure 1. Percentage of products with errors
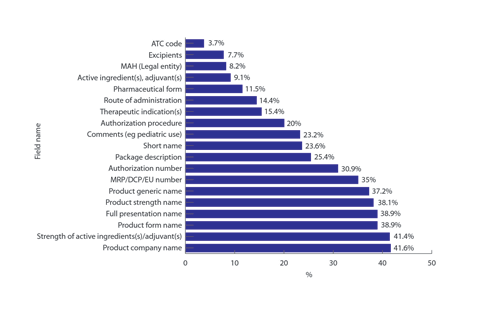
Areas of significant error were associated with:
- the full presentation name and associated name parts;
- the strength of active ingredients and adjuvants;
- authorization and procedure numbers;
- package descriptions; and
- pediatric use.
Many other fields had an error rate which the EMA considered to render the data unfit for the business purposes intended. Therefore significant parts of the guidance have been re-written to address many of the major quality concerns.
Modification of vocabularies
Significant quality issues also exist with the controlled vocabularies, particularly the ones for substances and organizations. Although this is not identified in detail in the announcements, the EMA is currently undertaking a significant clean-up of the substances vocabulary, identifying "master substances" and assigning other terms for these as synonyms. This will affect a large number of substances, which in turn will have an impact upon the EV codes used in the AMP records. For these changes the EMA will actually be updating the AMP records by assigning the new substance codes. Whilst this will be useful for EV Web users, for companies submitting via the EV Gateway (which accounts for about 60% of AMP records), substance codes will need to be updated in their own systems so that when future submissions are made to the EMA they will not be rejected because of substance codes that are no longer valid. The EMA will be producing a spread sheet of how it has remapped the substance codes but it will not produce a spread sheet for AMPs which have been affected as this is likely to affect a large percentage of them. Companies will need to check and modify records where necessary.
The vocabulary relating to organizations currently has a large number of duplicate MAHs included, where there are clearly only minor differences in name or address, eg Limited or Ltd, Avenue or Ave, etc. Companies will be expected to clean these up, make sure that the ones they want to keep are comprehensive and remove any duplicate records. This will affect many AMP records, which will need to have a different EV code assigned before the duplicate organization can be deleted.
SmPCs in local language for MRP/DCP products
Within the original requirements issued in September 20118, the EMA was requiring the local language SmPCs for all products authorized via the MRP and DCP plus all languages for centralized approval procedure (CAP) products. However, after industry lobbying, the agency refined the guidance9 so that it would accept the agreed English text for MRP/DCP products and only the English for CAP products. The just-issued guidance amends this to require the local language SmPC for MRP/DCP products. However, there may be circumstances when the agreed English text will be acceptable, as an interim, such as after a variation procedure when the local language SmPC is not available within the maintenance submission timeframe (see maintenance timeline below). In such cases, the local language SmPC would need to be submitted when it becomes available.
CAP products continue to require only the English SmPC.
Some items still not required
Industry will be pleased to learn that there is still no requirement for the structured substance information (SSI), which is deferred until the implementation of the ISO IDMP standards. Also deferred until then is the mandatory requirement for individual records to be submitted for each presentation included within a single marketing authorization. This remains voluntary although a large number of companies have already elected to submit these and it is recommended unofficially that they continue to do so.
Areas still lacking clarity
Much has been done to improve the clarity of the guidance, but there are still a few areas where greater clarity could be achieved. These include:
- how and when to supply translations for substance names and how to utilize them;
- whether records for Iceland, Norway and Lichtenstein (the non-EU members of the European Economic Area) are to be provided now; and
- definition of the maintenance clock-start for some specific procedures.
It is possible that these will be addressed in updates to the guidance or a reissue for frequently asked questions.
Maintenance of data
On 4 July 2012, just two days after the submission deadline, the EMA announced that the maintenance of the XEVMPD would be delayed until further notice10. However, a number of companies chose to ignore that guidance and have been updating their data during the interim.
For much of the intervening period, discussions have been taking place within the EMA and with industry associations regarding the processes, timelines and requirements for maintenance. Several proposals have come and gone before those outlined in the new guidance were adopted. A simplified process is to operate through to the implementation of IDMP in 2016. For most changes to the data, the update operation is to be used and the variation operation is never to be used. This is the main simplification in the process because the MAH will not have to determine which category of change something should be.
In principle, any changes to the data, or indeed some changes to the SmPC that do not affect the data (eg contra-indications, special warnings and precautions for use, undesirable effects) must be reported within 30 calendar days of when the change is approved (or can be implemented in the case of notifications). Guidance is provided regarding what the triggers are for the clock start. A key point is that for an MRP variation, it is the reference member state (RMS) approval that will trigger the clock start and this has a knock on impact as to whether the English or local language SmPC is able to be used.
A process has been designed to support the transfer of marketing authorization with action upon the old MAH to use "invalidate MA" and the new MAH to create a new record with a link made between the two via the old EV code. A similar process has been established for when there is a renewal in which the authorization number changes. Clarification is provided regarding the processes for suspensions, lifting of suspensions, revocations and withdrawal of marketing authorizations.
Companies will therefore need to establish processes to identify changes to all licenses, collection of data and the SmPC and provision to the EMA within 30 calendar days. This will be challenging for many companies and have a high cost in terms of the resources they need to manage.
Bringing the data up to date
In line with the EMA’s statement, the vast majority of companies have not kept their XEVMPD up to date. Even without new data requirements a significant proportion of the AMP records will now be out of date either because of regulatory changes to the data itself or because the SmPC has been updated without an impact upon the data. These changes now need to be submitted during the June to December 2014 window.
The following is a summary of what companies will have to do to bring their records up to date. It is not an exhaustive list as there are many subtle changes in the guidance that are too numerous to mention here, nor does it necessarily reflect an absolute order of activities.
Updating organizations
- identify the "definitive" organizations to be retained;
- correct the name and address details of definitive organizations;
- add SME information;
- submit updated organization records;
- change organization EV code for affected APM records – see updating authorized medicinal products; and
- nullify any now-redundant organizations once there are no product records that utilize the organization.
Updating authorized medicinal products
- undertake the transfer of marketing authorization process for any AMPs for which the regulatory activity is complete but the record still remains in XEVMPD. It is recommended that the EV codes are provided to the new MAHs so that they can take over responsibility for the product;
- withdraw any AMPs for which there is no longer a license;
- update the MAH EV code, if necessary, due to changes in the organization controlled vocabulary;
- update the substance EV code for any substances affected by the change to the substances controlled vocabulary (Gateway users only);
- add data for new mandatory fields;
- update any field where the data has changed since the initial submission (or latest update if some maintenance has been performed);
- update any field where data does not comply with the interpretation given in revised guidance;
- provide the latest version of the SmPC in local language for products approved under the national approval procedure (NAP)/MRP/DCP or English for CAP products,
- if a renewal has taken place where the authorization number has changed, then invalidate old record and insert new record; and
- delete any duplicated records with the reason for deletion included in the comments field (note: in the EMA's quality control pilot, the agency found that 5% of records appeared to be duplicates).
Inserting new authorizations/new records
Apart from those products that are newly authorized and so fall under the current 15 calendar-day reporting deadline, the EMA is recommending that the insertion of new authorizations/new records should be done after 16 June when the new schema and, hence the new fields, are available. If this is done before, records will have to be updated again post-June. The following should be addressed where applicable:
- following the transfer of marketing authorization newly transferred-in products should be added. The previous MAH should provide the EV code;
- if a renewal has taken place where the authorization number has changed, then a new record should be inserted once the old record has been invalidated. The previous EV code should be provided in the new record;
- records should be added for authorized products in Croatia (NAP/MRP/DCP) and the translations of substances provided (Croatia joined the EU in July 2013); and
- new authorizations received should continue to be submitted within 15 calendar days of notification of approval and the availability of the SmPC. However, for Gateway users this will be problematic until they have updated versions of software available that can process the new schema. In this case, the EMA is proposing a workaround whereby companies inform the agency of the authorization by email and of their submission plans to provide the records once they have the capability.
Technical changes necessary
Technical changes will be required for systems used for management and submission of the data. The EMA’s EV Web tool is being updated not only to support the new fields and picklists but also to change some "labels" so that some existing field names are displayed differently (eg authorization date becomes authorization/renewal date, MRP/DCP number becomes MRP/DCP/EMEA number). This is being done with the intention of improving clarity and hence the quality of the data provided. Improved search capabilities are anticipated and the EMA is also working on functionality to support the bulk updating of the same change to be applied to multiple records (eg the qualified person for pharmacovigilance (QPPV) code, organization code). To help companies check their data in XEVMPD, an export capability is being developed which will allow the export of up to 100 records at a time.
EV Gateway users (and EV-Post users) who generate their own XEVPRMs will need to update their software. At a minimum, they will need to handle the new fields and submit according to the new schema, but other functionality may be necessary, or useful, for example, to manage the process of updating multiple AMPs with the same data and to support some of the new maintenance processes. The scale of impact will vary from tool to tool but enhancement will need to be scheduled, implemented and tested before updated systems can be used for re-submission. This may be challenging for many companies to complete by the end of December, so careful assessment and planning will be necessary as soon as possible.
Conclusions
The new guidance will have significant impact upon industry, with many activities needed during 2014, plus more to support maintenance. Those companies directly involved in discussions through trade associations may have been aware of what was coming, but for most the requirements are likely to come as a surprise. In order to remain compliant, industry, plus its software and service providers, must initiate action quickly. Each company will need to conduct an impact assessment and determine the degree of change to records that will be necessary, establish project plans, allocate resources to undertake activities, establish new process, implement new versions of software and many more things besides. Does this sound familiar? Will companies face the similar challenges as in 2012 when the XEVMPD requirements were first introduced? The scale might not be quite so large but it is still very significant. The challenges may be very similar but at least this time everyone has the experience of XEVMPD the first time around to apply. It’s time to get started – again!
References
1. EMA press release, 31 January 2014, www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2014/01/news_detail_002014.jsp&mid=WC0b01ac058004d5c1
2. Commission Implementing Regulation (EU) No 520/2012 of 19 June 2012 on the performance of pharmacovigilance activities provided for in Regulation (EC) No 726/2004 of the European Parliament and of the Council and Directive 2001/83/EC of the European Parliament and of the Council, OJ, 20 June.2012, L159, pp 5-25, http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2012:159:0005:0025:EN:PDF
3. Legal Notice on the Implementation of Article 57(2) of Regulation (EC) No 726/2004: Electronic Submission of Information on Medicinal Products for Human Use by Marketing Authorisation Holders to the European Medicines Agency, 31 January 2014, EMA/505633/2011, www.ema.europa.eu/docs/en_GB/document_library/Other/2011/07/WC500108212.pdf
4. Reporting requirements for marketing-authorisation holders, EMA website accessed 6 January 2014, www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000594.jsp&mid=WC0b01ac058078fbe1
5. Detailed guidance on the electronic submission of information on medicinal products for human use by marketing-authorisation holders to the European Medicines Agency in accordance with Article 57(2), second subparagraph of Regulation (EC) No 726/2004, EMA website accessed 6 January 2014, www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/document_listing_000336.jsp&mid=WC0b01ac058079126c
6. Outlines of amendments to the XEVPRM schema and EVWEB Labels Technical Specifications, 31 January 2014, EMA/21088/2014, www.ema.europa.eu/docs/en_GB/document_library/Other/2014/01/WC500160459.pdf
7. Proposal for a regulation of the European Parliament and of the Council on fees payable to the European Medicines Agency for the conduct of pharmacovigilance activities in respect of medicinal products for human use, 26 June 2013, COM(2013) 472 final, 2013/0222 (COD), http://ec.europa.eu/health/files/fees_2013/comm_native_com_2013_472_proposal_for_a_regulation_en.pdf
8. Detailed guidance on the electronic submission of information on medicinal products for human use by marketing authorisation holders to the European Medicines Agency in accordance with Article 57(2), second subparagraph of Regulation (EC) No. 726/2004 (Chapters 1-6) (Version 2.0) (NB September 2011 version no longer available on line)
9. Detailed guidance on the electronic submission of information on medicinal products for human use by marketing authorisation holders to the European Medicines Agency in accordance with Article 57(2), second subparagraph of Regulation (EC) No 726/2004 (Chapters 1-6) (Version 3), (NB March 2012 version no longer available on line)
10. EMA press release, 4 July 2012, www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2012/07/news_detail_001558.jsp&mid=WC0b01ac058004d5c1
Andrew P Marr, PhD, is managing director of Marr Consultancy Ltd, a consultancy firm based in Knebworth, UK, that provides services to the pharmaceutical industry, software vendors and service providers to the industry, specializing in the extended Eudravigilance Medicinal Product Dictionary (XEVMPD) and ISO Identification of Medicinal Products (IDMP). Email: [email protected].