The Commercial Impact of REMS Relief: Nplate/Promacta Sales Spike After Risk Management Changes
This article was originally published in RPM Report

Executive Summary
Nplate and Promacta aren’t the most important products sold by Amgen and GSK. But they are among the fastest growing in 2012—thanks to a decision by FDA to relax post-market controls at the end of 2011. The change underscores the case for a different approach to REMS in oncology, and may point the way forward for broader use of the new tools in a less intrusive way.
Promacta isn’t the type of product that usually gets mentioned by the CEO of a global pharmaceutical company during a quarterly earnings call. At less than $200 million in annual sales, Promacta (eltrombopag), approved for thrombocytopenia, is barely a rounding error in the performance of GlaxoSmithKline PLC .
But the product came up not once, but twice during GSK’s third quarter call. The first mention came during the scripted run-down of new product performance during the quarter, where Promacta’s 50% growth rate was cited by CEO Andrew Witty in a recap of the company’s “newer products in the specialty franchise.”
And Witty cited it again in Q&A, when he was asked to expand on his enthusiasm for the newer specialty therapies, given their relatively small impact on the bottom line.
Witty reiterated Promacta’s growth—this time noting that it is almost 60% year-to-date—as one in a series of smaller products with “a very punchy growth rate.”
“And what we've seen is that gradually those products are beginning to turn into something pretty chunky,” Witty added. However, he acknowledged, “they're always going to be limited by the fact that they are in specialist marketplaces.”
You might think that Promacta’s rapid growth rate would be bad news for the other product FDA approved for thrombocytopenia in 2008, Amgen Inc.’s Nplate (romiplostim). But no: Nplate sales are doing just fine, up more than 30% through the first nine months of 2012.
In fact, the combined growth rate of the two thrombocytopenia therapies is just over 40% in the US so far this year. Not bad for a pair of four-year old products.
So is there a sudden surge in thrombocytopenia driving the demand for the two drugs?
Probably not.
Rather, the growth spurt likely has much more to do with a regulatory action taken by the Food & Drug Administration in December 2011. That was when FDA agreed with the two sponsors that their programs could be amended to eliminate onerous registration and certification requirements—known as Elements to Assure Safe Use (ETASU) – and replaced by a much less demanding communication program to be administered in conjunction with professional societies representing oncologists and hematologists who use the products.
The resulting increase in sales speaks for itself. (See Exhibit 1.)
Exhibit 1
A Step-Up in 2012
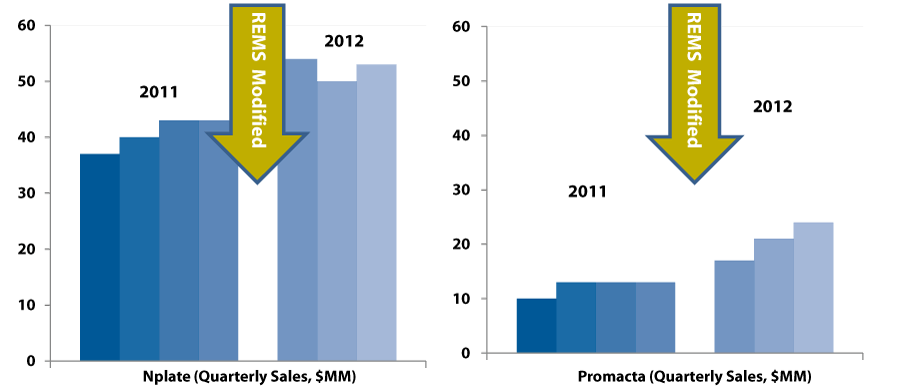
SOURCE: The RPM Report, company reports
A Head-to-Head Battle Becomes Fight Against Common Enemy
From the time of launch, the head-to-head battle between GSK and Amgen in the thrombocytopenia category looked like an important case study in how sponsors would adapt to the REMS era. (Also see "Living With REMS: The New Regulatory Model Meets Commercial Reality" - Pink Sheet, 1 May, 2009.)
Promacta and Nplate were among the first products approved with the most restrictive type of post-marketing controls. The fact that they had the same indication in a small market made the head-to-head nature of the competition paramount—as did the fact that other aspects of the two therapies are very different (most notably, Promacta is an oral therapy while Nplate is injectable).
Two things quickly became apparent.
First, Nplate got the lion’s share of the market—roughly 75% of the US sales by dollar volume. The head-to-head fight wasn’t really that close.
But, second, neither therapy reached its potential, largely because the REMS obligations were so onerous that hematologists opted not to use either therapy (and may, perversely, have been encouraged to use an unapproved therapy—Rituxan—which did not come burdened with a REMS.)
In other words, what looked like a head-to-head fight to capture a relatively small new market instead became a fight against a common enemy: the onerous REMS obligations retarding uptake of both products.
Now that the REMS requirements have been lifted, Promacta and Nplate become a case study of another kind: the impact that easing post-market controls can have on commercial performance.
In this case, the transition from strict distribution controls to a softer-touch model has meant a 40% increase in the size of the market, almost overnight. Call it “REMS relief.”
Beneficiaries of a Broader REMS Rollback
The regulatory change didn’t happen in isolation.
FDA made the change simultaneously for both sponsors, and in the context of a broader rethinking of the role of REMS in the health care system—including a focused discussion about the unique aspects of REMS in oncology.
That discussion began in part because of Nplate and Promacta: the restricted distribution programs were really the introduction of REMS to the oncology community. From the first, providers found the programs intrusive and burdensome, and many voted with their feet—or, perhaps, their prescription pads—and opted for off-label Rituxan rather than follow the program.
The negative reaction to the REMS was somewhat surprising, considering that hematologists already participate in one of the model programs upon which REMS are based, the Celgene Corp. STEPS program for Revlimid.
However, that underscores the main message from providers in general and oncologists in particular: it isn’t so much the specific burdens associated with a given program as it is the proliferation of different programs each of which requires separate time and care to comply with.
Still, the impact of REMS in oncology weren’t that big in 2008 and 2009, and the concerns about the thrombocytopenia REMS remained in the background.
That changed in 2010. The triggering event for the reconsideration of REMS in oncology came with FDA’s imposition of a registration program for prescribers of erythropoietin stimulating agents (Amgen’s Aranesp and Johnson & Johnson’s Procrit.) (Also see "The Rebirth of A Niche Product: Three Silver Linings on the ESA REMS" - Pink Sheet, 1 Feb, 2010.)
Now every oncologist was exposed to the new tools, and—like most providers—they didn’t like what they saw.
But oncologists aren’t just any provider group, and their concerns clearly resonated inside the agency. The American Society for Clinical Oncology responded quickly to the ESA REMS, and was given the lead-off slot in FDA’s July 2010 stakeholder meeting to discuss the impact of the programs on the health care system.
And ASCO was successful in making the case that cancer merits special attention in the context of that conversation. After the stakeholder meeting, FDA agreed to participate in a workshop focused specifically on the role of REMS in oncology. (Also see "Carving REMS Out of Oncology? FDA, ASCO Plan Follow-Up Workshop " - Pink Sheet, 1 Dec, 2010.)
That workshop (invitation only) took place in July 2011, and a summary has just been published in ASCO’s Journal of Oncology Practice. (The article was published online![]() October 30.)
October 30.)
Oncology is Special, but REMS Issues are Not
The key messages from the workshop were not surprising: case-studies helped quantify the administrative burdens imposed by REMS on practices and the sense that the added burden does not add much (if any) value to the management of patients.
The published summary, however, suggests a somewhat unexpected twist: while oncologists and FDA clearly are in agreement that the issues of cancer care are unique, the solutions to address the impact of REMS really are not any different than for every other health care sector.
The workshop focused on the need for standardization of REMS, input from practitioners into REMS design, and carefully choosing an approach that minimizes the burden on providers and integrates into existing systems as seamlessly as possible.
Those are the same broad themes of stakeholder input to FDA across the provider spectrum. (Also see "REMS 2.0: FDA Refining New Drug Safety Tools" - Pink Sheet, 1 Dec, 2010.)
Indeed, so consistent has been the feedback to FDA that the agency has already initiated changes that align with the priorities identified by the ASCO-organized workshop. (Also see "FDAAA Impact Analysis (Year 4): The REMS Retreat Continues - For Now" - Pink Sheet, 29 May, 2012.)
As a result, the workshop summary has a somewhat anticlimactic conclusion: the authors note that there have been a lot of changes since the workshop was held, including the elimination of the routine expectation that a MedGuide requirement would trigger a REMS and the integration of REMS standardization efforts into the reauthorization of the Prescription Drug User Fee Act.
“The FDA has proposed taking meaningful steps in the context of PDUFA reauthorization to standardize and integrate REMS into the health care system,” the article concludes. “The provider community and professional societies are eager to build on this opportunity.”
Toward a Tiered Model?
However, it would be a mistake to say the ASCO initiative had no impact beyond contributing one prominent voice to the chorus of calls for changes in the implementation of REMS.
In fact, the workshop process itself may serve as a model for future work by FDA on gathering stakeholder input.
“Ongoing dialogue between stakeholders is imperative to the future of drug safety management,” the summary notes. “This ASCO workshop, which included stakeholders from across the health care landscape, suggests subsequent similar forums and working groups may be an effective way to chart a path forward for REMS. Smaller groups could focus on specific aspects of REMS and explore other risk management options, including the use of informatics to improve education and assessments and how to leverage best practices currently in place in order to refine REMS programs.”
In addition, the workshop includes discussion of a “tiered” approach to REMS in oncology that may emerge as good working model for decision making in applying REMS in cancer care—and perhaps more broadly.
The three tiered approach would help FDA classify risks as “typical” for oncology and therefore not requiring a REMS; different from common toxicity and therefore an appropriate candidate for a communication plan; and finally risks so great that marketing is not possible without mitigation strategies that deliver “real-time” information to providers (and would likely be an ETASU REMS). (See Exhibit 2.)
Exhibit 2
Decision Tool for an Alternative REMS Approach for Hematology/Oncology Products
An excerpt from the summary of a July 2011 workshop on REMS in oncology, organized by ASCO and other provider groups.
|
The workshop organizers presented a suggested approach that involves a decision tool to guide decision making about REMS development and implementation for hematology/oncology products. The tool would place each drug into one of three possible levels: |
|
Level 1 would apply to drugs with toxicities similar to those already well managed in clinical practice; this would include the majority of oncology drugs. Level 1 drugs would not need an REMS. The FDA and the drug manufacturer would provide standard information, but no changes to standard practice would be required. |
|
Level 2 would apply to drugs that carry risks different from common toxicities. These toxicities, however, are manageable given appropriate evidence-based information because of safety measures already common to clinical practice. A REMS program for a level 2 drug would involve a core set of safety information developed by the FDA and the drug manufacturer. The oncology community could then assist in further developing and disseminating important information through various education and communication vehicles. If applicable, laboratory testing or other safe-use procedures could be incorporated into an EHR. |
|
Level 3 would apply to drugs that would not be approved in the absence of risk mitigation strategies. Level 3 drugs carry serious, unique risks that should be highlighted for providers and patients and cannot easily be managed by existing practice measures. This could also apply to drugs that carry a specific risk not familiar to a given provider population. REMS programs to manage these drugs should integrate drug safety information in real time in a way that assists patient and provider decision making. Examples could include evidence-based guidelines created by professional organizations, decision aids embedded in EHRs, and quality measures to assess comprehension and implementation into practice. |
Assessment of Risk Evaluation & Mitigation Straegies in Oncology, Journal of Oncology Practice, Oct. 30, 2012
The workshop is also a model of a different kind, since it clearly also served as a forum to work through specific issues relevant to the Nplate and Promacta REMS. The key change approved by FDA involves replacing a restricted distribution program with a communication plan—a move from “tier 3” to “tier 2” on the scale discussed at the workshop.
The newly approved REMS also emphasizes the interaction with provider organizations to help disseminate the message—an important topic of discussion during the July 2011 workshop.
“The responsibility to implement safety standards in practice can best be fulfilled by providers,” the summary article states. “Provider and patient organizations have a variety of tools to increase patient safety and assess quality measures….In addition, professional societies are best equipped to work with health information technology developers to create systems to prompt providers and patients about safety concerns.”
Based on FDA documents discussing the new REMS requirements, the formal process to modify the two REMS began in late 2010—at the same time ASCO and FDA were organizing the workshop.
It also coincided with FDA’s consideration of a REMS for Bristol-Myers Squibb Co.’s melanoma therapyYervoy. As part of that review, FDA considered an ETASU REMS, but after internal deliberation agreed on a communication-plan-only REMS. That approval came in March 2011.
The Nplate/Promacta REMS was one of the case studies presented by FDA officials during the July 27 workshop itself. In fact, FDA sent “REMS advice letters” to both sponsors on the eve of the meeting. (See Exhibit 3.)
And then the ETASU limitations were lifted in December.
Exhibit 3
Lifecycle of a REMS
Key dates for Nplate and Promacta
|
Aug. 22, 2008 |
Nplate Approved with Restrictive REMS |
|
Nov. 20, 2008 |
Promacta Approved With Restrictive REMS |
|
Oct. 25, 2010 |
Richard Pazdur discloses plans for REMS workshop with ASCO |
|
Nov. 12, 2010 |
FDA issues “REMS Modification Notification” letters for Promacta and Nplate |
|
Jan. 11, 2011 |
Promacta Supplemental NDA Submitted, including REMS modifications |
|
Feb. 11, 2011 |
GSK/FDA teleconference on proposed REMS modifications |
|
Feb. 24, 2011 |
Amgen/FDA teleconference on proposed REMS modifications |
|
March 31, 2011 |
Amgen files Supplemental NDA including REMS modifications |
|
July 25, 2011 |
FDA issues “REMS advice letters” to Amgen and GSK |
|
July 27, 2011 |
ASCO hosts Oncology REMS workshop |
|
Dec. 6, 2011 |
FDA approves new REMS for Nplate and Promacta |
Source: The RPM Report, FDA
Tradeoff for Industry: Less Information, More Sales
As emphasized by the workshop participants, the new requirements emphasize the role of the manufacturer in providing tools, but relying heavily on provider groups to deliver the messages.
Specifically, the REMS requires Amgen and GSK to send a “Dear Professional Society” Letter in addition to the more typical “Dear Healthcare Provider” Letter. Those letters serve as an alert that restricted distribution is no longer required for the products, and also as a reminder of the serious adverse events that need to be considered in using the therapies.
That is a significant reduction in the level of control over the products; the original REMS included formal registration of physicians, pharmacies and patients, with an explicit requirement for pharmacy verification of eligibility before dispensing the therapies.
But there is a trade-off in moving to a “lighter touch” REMS, which is noted in the discussion of the ASCO workshop.
In the introduction to the summary, the authors note the “unintended consequences” of REMS include “disclosure of patient information to drug manufacturers.”
“Implementation of the most restrictive REMS elements functionally inserts manufacturers into the patient-provider relationship for drug acquisition and safety monitoring,” the article notes. “Some REMS programs include a requirement for patients and providers to disclose confidential medical information to gain access to a marketed drug.”
The summary notes that those requirements can pose challenges for manufacturers as well, since “access to information on prescribing patterns and involvement in REMS-required patient and provider monitoring may run counter to federal and state privacy laws.”
“These activities reside better with providers than with manufacturers,” the workshop summary states. “Demonstrating steps that build safety mitigation into daily practice should be sufficient to avoid provision of patient information to manufacturers."
For manufacturers, the goal of REMS reforms is not so much “to avoid” collecting information on their customers, but rather to increase the customer base by limiting barriers to access. In the case of Nplate and Promacta, it was relatively clear that the collection of information was indeed restraining sales—and the release of the REMS has shown that the impact was significant.
But it is interesting that both manufacturers didn’t abandon every element of the REMS that FDA waived.
In the case of Amgen, the company announced the REMS modifications in a press release, and included a prominent notice about its oncology patient support program, First Step. The Nplate website continues to offer patient registration, with the My Nplate patient support center. The lure of financial and logistical assistance has replaced the mandate to register to get the medication, but Amgen is still understandably eager to link directly to end customers.
For Promacta, GSK does not maintain a prominent patient-focused presence on the web. However, the company did maintain a restricted pharmacy network for distribution of Promacta even after the REMS was lifted.
That suggests that restricted distribution per se isn’t the problem for manufacturers; it is only when the restrictions don’t align with the expectations of providers that they become an issue.
In oncology, that likely means communicaton plans rather than ETASU REMS will be the norm, with relationships between manufacturers and medical societies providing the backbone of successful communication. That lesson is likely to resonate even after the sales curve flattens again for Promacta and Nplate.