FDA Pushes Back Against “Living” Benefit-Risk Assessment Management Plan
Executive Summary
The Institute of Medicine urges the agency to create a publicly available document setting forth benefits and risks of drugs, with periodic updates, but FDA says it has no extra resources to handle such an effort.
FDA agreed during user fee negotiations to put in place a systematic approach for benefit-risk assessment of drugs and for communicating information gathered after market launch, but the agency views a “living” benefit-risk assessment management plan as a step too far given the current workload.
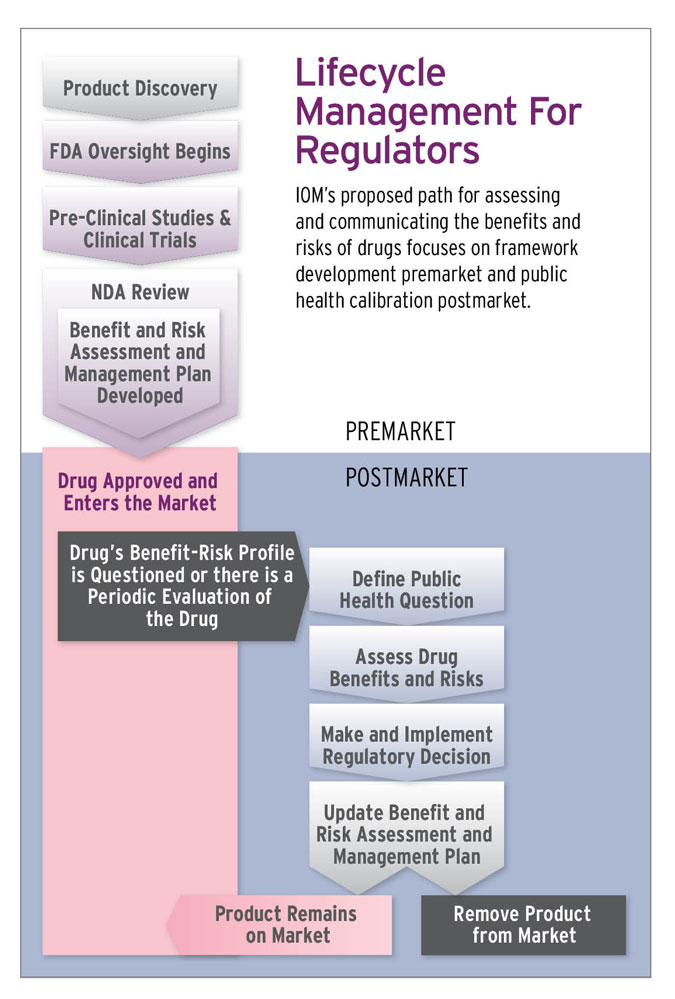
The Institute of Medicine released a report May 1 recommending the agency create for each new and already approved drug a publicly available and understandable benefit-risk assessment management plan (BRAMP), which would be updated as new data comes in, providing patients with a single, comprehensive source of information about the benefits and risks of a medication throughout its market life.
FDA accepts “the general concept of enabling the public to be able to clearly monitor relevant safety issues for all drugs,” the agency said in a same-day statement. “However, we believe it would be very challenging to implement this recommendation within our current resources without seriously compromising other critical regulatory activities.”
IOM’s call for more information-sharing also could run into legal hurdles, FDA said. “Some of the recommended changes in disclosure policies would require changes to existing laws and regulations.”
European regulators have some experience with annual reviews of products’ benefit-risk balance – but even on a going-forward basis only, it would require significant additional resources for FDA to implement.
BRAMP Updated As FDA Evaluates New Data
The BRAMP envisioned by the 12-member committee convened by the IOM would be updated at pre-specified times and whenever FDA reevaluates a drug’s benefit-risk profile. For new drugs, the document should be initiated during the drug-approval phase, according to the committee’s report, “Ethical and Scientific Issues in Studying the Safety of Approved Drugs.”
An important component of the BRAMP would be an FDA explanation of the rationale for its regulatory decisions, including requirements for post-market research or a Risk Evaluation and Mitigation Strategy, and an evaluation of the effectiveness of those decisions.
Industry reaction to more FDA information sharing has been mixed. During development of FDA’s transparency policy, drug makers sought more information on why FDA makes the decisions it does (Also see "PhRMA Wants FDA To Explain When Decisions Depart From Committee Votes" - Pink Sheet, 29 Jun, 2009.). More recently, the Biotechnology Industry Organization called on FDA to disclose in “complete response” letters why a product’s risks outweigh its benefits as part of its “big ideas” for regulatory reform (Also see "BIO’s "Big Ideas" For FDA Reform Are Too Big For PDUFA" - Pink Sheet, 4 Jul, 2011.).
However, more information sharing with the public can be muddied by confidentiality issues. An FDA transparency task force proposal to release redacted “complete response” and refuse-to-file letters raised questions from BIO about what information would be removed (Also see "FDA Transparency Initiative: Disclosure Rules Could Expand, Then Be Harmonized" - Pink Sheet, 24 May, 2010.).
The institute suggests FDA convene a working group of industry, patient and consumer representatives to find a way to disclose data from trials and studies while balancing public health, privacy and proprietary interests.
For now, IOM suggests the BRAMP include a description of any public health questions; benefits and risks related to that issue; key stakeholder input specific to each question; a schedule for future benefit-risk assessment; plans for, and results of, evaluating the effectiveness of any regulatory decisions or actions, and a discussion of disagreements about available evidence during regulatory decision-making. Design details of post-market surveillance studies would be included.
IOM outlines a three-stage regulatory decision framework that can be utilized throughout a drug’s life cycle to assess drug safety, including as part of post-market benefit-risk updates. The framework involves defining the public health questions, assessing benefits and risks and making and implementing regulatory decisions.
Bringing consistency and patient values into benefit-risk decisions is one of the goals of the user fee agreement negotiated by industry and FDA (Also see "PDUFA V Agreement Set In Four Areas, But Broad FDA/Industry Talks Continue" - Pink Sheet, 7 Mar, 2011.).
The Pharmaceutical Research and Manufacturers of America hosted a working group on the nuts and bolts of benefit-risk analysis that developed a structured and systematic method to consider the information necessary for weighing a product’s benefits and risks (Also see "PhRMA Testing Pilot Framework For Structured Benefit-Risk Assessment" - Pink Sheet, 12 Sep, 2011.). PhRMA moved work on the Benefit-Risk Action Team framework to the independent Centre for Innovation in Regulatory Science Ltd., which is seeking to harmonize benefit-risk assessment across companies and regulatory agencies (Also see "Benefit-Risk Assessment Framework Moves Toward Global Harmonization" - Pink Sheet, 30 Jan, 2012.).
Recent draft FDA guidance highlighted that benefits, as well as risks, are part of the drug approval equation. The guidance asks industry to provide evolving efficacy information in Periodic Benefit-Risk Evaluation Reports (Also see "Post-Market Safety Updates Replaced By “Periodic Benefit-Risk Evaluation Reports”" - Pink Sheet, 10 Apr, 2012.).
Criteria For Post-Market Studies
IOM says FDA’s safety efforts would improve if the agency delineated the risk factors that could trigger post-market studies. When these occur, the agency should make public the rationale for either requiring a study, or not requiring a study “if there is a compelling argument against it.”
Factors Indicating Uncertainty About Benefit-Risk
- Approval using several surrogate endpoints that provide conflicting evidence about likely health outcomes
- First-in-class approval based on surrogate endpoints employed for a different drug class
- A drug with a worrisome safety profile that could affect a large number of people, with a severe side effect or a strong biological rationale for a particular side effect
- A drug is expected to have a different benefit-risk profile in a subgroup under real-world conditions
- A drug is in a class that has a previously identified, substantial safety risk
- Evidence of lack of benefit in the target population or identifiable subgroup emerges post-marketing
If a study is called for, IOM says a randomized controlled trial should be the option only when an observational study cannot obtain the needed information and the RCT can get data within the necessary timeframe and is ethically acceptable.
In the post-market setting, there are heightened informed consent concerns, IOM points out, and recommends FDA issue guidance for interpreting disclosure and informed consent requirements. The institute provided an early review of the ethical issues in post-market trials in a report released in the lead-up to an advisory committee to discuss halting the TIDE study of GlaxoSmithKline Inc.’s Avandia (rosiglitazone) and Takeda Pharmaceutical Co. Ltd.’s Actos (pioglitazone) (Also see "IoM Report On Postmarketing Study Ethics Could Drive Broad Overhaul Of Informed Consent" - Pink Sheet, 12 Jul, 2010.).
IOM calls for early delineation of the potential public health questions, as well as key standards and definitions for studies and planning for meta-analyses.
FDA already encourages early planning for meta-analyses – for example, in its guidance on assessing cardiovascular risk in diabetes drugs. Findings from a meta-analysis of Phase II and III trials can rule out the need for a CV outcomes trial or delay it until after approval. FDA is considering a similar approach for obesity drugs (Also see "CV Risk Assessment For All Obesity Drugs Seems Inevitable Pre-Approval Requirement" - Pink Sheet, 29 Mar, 2012.).
Because surrogate endpoints increase the uncertainty of a drug’s benefits and risks, IOM says FDA should maintain and annually update a list of surrogate endpoints used in drug approval and explain the rationale for their use. The list should include post-market evidence for their correlation to the health outcomes of interest and list their successes and failures “so that for each major drug class, the regulatory science related to approval methods can be modified and improved,” IOM says.
FDA has worked on an inventory of surrogate endpoints under the auspices of the Critical Path initiative (Also see "FDA’s Progress On Biomarkers: Two Steps Forward, One Step Back?" - Pink Sheet, 28 Nov, 2005.).
So Many Things To Do
The IOM report gives FDA advice on a wide range of other activities relating to review practices.
- Guidance and review processes should ensure that observational studies with high internal validity are given appropriate weight when evaluating harm and transportability is given the same emphasis as bias and other errors in assessing weight-of-evidence.
- FDA should have adequate expertise in Bayesian analyses, as well as in relevant frequentist and causal inference methods, and develop guidance on using Bayesian methods to prepare a benefit-risk profile.
- FDA should present data and analyses so independent analysis can reproduce the results and understand how they were derived.
- FDA should issue guidance on the design and conduct of non-inferiority post-market safety trials and use the observed-effect estimate and confidence interval as the basis for decision-making, not the binary non-inferiority verdict.